作者:姜翰昕 郭錦龍 / 中研院原分所博士後研究員 臺灣大學材料系副教授
隨著電腦科技的不斷進步,我們可預期直接使用量子力學理論預測材料性質的第一原理計算(first-principles calculations)在未來的材料系統設計中的角色將會越來越吃重。維也納大學開發的VASP (Vienna ab initio Simulation)軟體由於功能強大且計算效率高,目前已經是進行此方面研究時最被廣泛使用的計算程式之一。我們計畫用三篇文章簡介VASP在計中操作環境下的操作,並且運用實際例子示範如何運用VASP進行研究。作為系列作的第一篇,在本文中我們演示VASP在計中環境下的基本操作以及進行最單純的結構最適化的步驟,並同時介紹如何判讀VASP的基本輸出檔。
前言
隨著科技的進步,可用於能量儲存或電子元件的新材料開發也越來越急迫。在開發材料時,運用第一原理材料模擬計算的技術可幫助預測系統的微結構以及許多重要性質,如相穩定性、原子動力學、化學反應性、光電性質等。若我們能對這些性質進行適當的預測,將可省去在材料發展過程中過多無謂的試誤過程,降低開發時所需要的時間與金錢成本。然而,過去由於電腦技術的限制,雖然科學家們已開發出多種材料計算模擬的理論,材料模擬技術尚無法在產業上進行實際應用。近年來,隨著電腦技術的快速進步,材料模擬在當今的產業鏈中的角色漸見吃重,而在學術界相關的研究亦如雨後春筍般蓬勃發展。為了跟上這股潮流,吾人必須要先能夠掌握現今常見材料模擬計算工具的背後原理與使用方法。
在金屬或半導體的塊材系統的模擬計算相關研究中,由維也納大學開發的量子物理模擬計算軟體Vienna Ab initio Simulation Package (VASP)可說是最被廣泛使用的軟體之一。此軟體的計算根基於能將多電子系統之波函數簡化為以電子密度分布的泛函表示的單電子系統的密度泛函理論(density functional theory, DFT),而在計算中電子密度的波函數則是以平面波的數學形式展開。此外,VASP的計算也套用了布洛赫定理(Bloch’s theorem)。這些特點使得此軟體相當適合處理帶有週期性邊界條件(periodic boundary condition, PBC)的塊材系統的問題。此外,由於此軟體的平行化效能相當優秀,若有適當的硬體設備配合也可對較大的系統進行有效率的模擬計算。由於以上因素,近年來無論是傳統的金屬與結構陶瓷材料的改良,或是新興的能源材料或電子元件材料的設計,VASP的計算預測皆扮演相當重要的角色。在本次的電子報中,我們將會以實例操作的方式簡介VASP最基本的使用法,而這些內容將可做為熟悉此工具的第一塊敲門磚。以下我們將介紹一些此軟體的基本使用方式。
二、基本操作:結構最適化(structural optimization)
二-1 運用VASP進行結構最適化之理論基礎
在此練習中,我們將會演示如何透過結構最適化過程得到結晶系統之晶格常數(lattice constant)與完美晶格內之原子位置。結構最適化是一個透過改變系統中的原子位置和晶格形而使得系統內原子間的相互作用力最小化的過程,而將應力最小化後得到的結構即為材料的平衡結構。此計算是一切材料計算的根本,因為在材料計算上有意義的結果其值必須要是由平衡結構中取得。在VASP的計算中,原子間的作用力是透過DFT加以預測。根據DFT的原理,如果我們可以藉由計算中得到合適的電子密度分布函數ρ_0 (r ? ),我們即可藉由此電子密度分布計算系統的性質。以下流程圖簡單敘述了VASP利用DFT進行「自洽計算(self-consistent calculation)」得到ρ_0 (r ? )的計算流程:
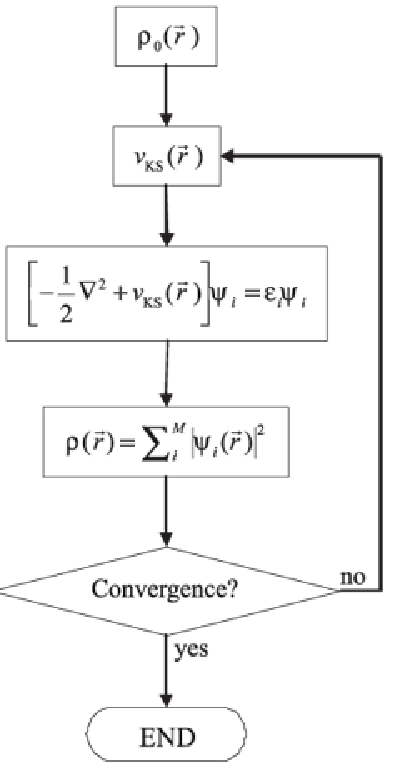
(source: J. Braz., Chem. Soc. vol.20 no.7, Sao Paulo (2009))
在本電子報中,我們將示範結晶矽與二氧化矽的結構最適化過程與對應的輸入與輸出檔,以使讀者更容易了解VASP的使用方式。
二-2 實際操作:結晶矽的結構最適化
在此計算中,VASP需要以下基本輸入檔案:INCAR、POSCAR、POTCAR、KPOINTS。而以下將討論這些檔案的內容 INCAR: 進行計算工作時的參數設定 下面為本次計算中所使用的INCAR檔案,包含了跟結構最適化相關的所有參數。
ISIF = 3
ENCUT = 500
NSW = 500
IBRION = 2
ISPIN = 2
EDIFF = 1E-4
EDIFFG = -5E-2
若想要更加理解這些參數背後的意義,可直接查閱VASP的線上操作手冊,其內容相當詳細(http://cms.mpi.univie.ac.at/vasp/vasp/vasp.html),在此將只簡述計算中列出的部分參數。ISIF=3代表在此結構最適化計算過程中系統的體積及對稱性是可以被改變的。ENCUT = 500則代表將波函數以平面波基底展開時各平面波基底對應的能量最高可到500 eV。IBRION = 2表示在結構最適化過程中系統的能量收斂過程是藉由共軛梯度法(conjugate gradient method)進行。ISPIN = 2代表計算中系統的波函數有考慮電子自旋。EDIFF為電子進行自洽計算之能量收斂定義範圍,而在本計算中此值為10-4 eV。EDIFFG則是原子間作用力的收斂值,此指令表示結構最適化的過程將會在每顆原子上的受力都小於0.05 eV/A時結束。
POSCAR: 欲計算之系統的晶格形狀及原子位置 下圖為此計算中使用的POSCAR檔案。在此檔案中,我們給予Si晶體結構之晶格常數的初猜值為5.3 A。
Si
1.0
0 2.65 2.65
2.65 0 2.65
2.65 2.65 0
Si
2
Direct
0.000000000 0.000000000 0.000000000
0.250000000 0.250000000 0.250000000
在此POSCAR檔案中,第一行為自行訂定的檔案標示,對計算結果無影響。第二至第五行定義了計算系統的三軸長度以及對稱性。由於Si的晶體結構具有Fd-3m之對稱性,其三軸向量為面心立方晶體(FCC)之展開基底。第六至七行定義系統內的原子種類及個數,而在本例中Si的原胞含有二顆原子。第八行及之後則是定義原子在晶胞內的位置,其中第八行的Direct代表以下的原子座標皆由在晶格內的相對座標表示,而九至十行的兩列數字代表原胞內的二顆原子的位置。
KPOINTS: 布洛赫定理中倒空間「布里淵區」(Brillouin zone)之k-point的取樣
下圖為本計算使用的KPOINTS檔案:
Si_primitive
0
Monkhorst
10 10 10
0 0 0
在此檔案中,第一行為執行計算工作的名稱,而這行內容可以自訂,不影響實驗結果。第二行的”0”代表由程式自動產生k-point取樣格點。第三行為取樣方式,我們選用Monkhorst-Pack的格點分割法。第四行定義取樣格點的密度。由於此系統晶胞極小且原子數只有二顆,其k-point必須要相當密集。在此範例中我們將布里淵區內所有方向皆分為10個格點。最後一行則代表我們在取樣時基準點定在原點。通常若無特殊需求該值並不會有所更動。
POTCAR: 原子周圍電子的潛勢與原子位置的關係
與前三者不同,POTCAR的檔案難以由使用者手動設定。不過VASP軟體本身會提供現成可使用的檔案,而且由於VASP製作相關參數的過程相當嚴謹,直接使用軟體提供的檔案可做出可信度高的結果。在h71上,POTCAR的檔案/opt/vasp/vasp_src/資料夾中找到:
[chinlung@h71 Geo_Opt]$ ls /opt/vasp/vasp_src/
bench.Hg.tar.gz potUSPP_GGA.tar.gz vasp-download-2015-11/
patch/ potUSPP_LDA.tar.gz vasp-download-2016-08
potpaw_GGA.tar.gz vasp.5.3.5.tar.gz vdw_kernel.bindat.big_endian.gz
potpaw_LDA.52.tar.gz vasp.5.4.1.tar.gz wannier90-2.0.0.tar.gz
potpaw_PBE.52.tar.gz vasp.5.lib.tar.gz
在本範例中,我們採用GGA-PW91做為exchange-correlation energy的推估方法,而相對應的壓縮包為potpaw_GGA.tar.gz。將其複製並解壓縮後,我們找出Si的資料夾,並將資料夾內POTCAR.Z進行解壓縮,即可得到Si的POTCAR。POTCAR檔的前幾行如下:
PAW_GGA Si 05Jan2001
4.00000000000000000
parameters from PSCTR are:
VRHFIN =Si: s2p2
LEXCH = 91
EATOM = 103.3076 eV, 7.5929 Ry
從這幾行中,我們可以得知在此POTCAR中每顆Si原子上有4個價電子,其組態為2個s電子加上2個p電子。同時,價電子間的exchange-correlation energy是由PW91 (LEXCH = 91)的計算法加以描述。
在本例子中,我們使用的VASP版本為5.3.5,而此版本使用計中h71主機上提供的執行批次檔。該檔案的位置在位置在/opt/vasp/vasp5/vasp5.3.5/pbs.sh,其內容如下:
### NOTE!! This PBS-script just used to vasp in h71-cluster
#!/bin/bash
###
### If you want to change the run-nodes-number , you can change "nodes=[nodes]".
###PBS -l nodes=6:ppn=12 -l mem=10gb -l vmem=10gb
#PBS -l nodes=1:ppn=20
#PBS -N vasp5
usecpu=$(cat $PBS_NODEFILE |wc -l)
module load openmpi-intel
cd $PBS_O_WORKDIR
export OMP_NUM_THREADS=1
export MKL_NUM_THREADS=1
### For gamma
#/opt/vasp/vasp5/vasp5.3.5/vasp-gamma/vasp
### For noncollinear
#/opt/vasp/vasp5/vasp5.3.5/vasp-noncollinear/vasp
### For default
mpirun -np $usecpu /opt/vasp/vasp5/vasp5.3.5/vasp-standard/vasp
執行計算時,我們僅需要執行qsub指令(qsub pbs.sh),即可將計算工作送至節點。完成計算後,資料夾內有許多新生成的輸出檔:
CHG DOSCAR INCAR OSZICAR POSCAR Si_P.e53097 Si_P.o53097 WAVECAR
CHGCAR EIGENVAL jobvasp-5.35.sh OUTCAR POTCAR Si_P.e53098 Si_P.o53098
XDATCAR CONTCAR IBZKPT KPOINTS PCDAT Si_P.e53065 Si_P.o53065 vasprun.xml
其中,與結構最適化較為相關的輸出檔為CONTCAR、OSZICAR、以及OUTCAR。
CONTCAR
Si
1.00000000000000
0.0000000000000000 2.7342938815236502 2.7342938815236502
2.7342938815236502 0.0000000000000000 2.7342938815236502
2.7342938815236502 2.7342938815236502 0.0000000000000000
Si
2
Direct
-0.0000000000000000 -0.0000000000000000 -0.0000000000000000
0.2500000000000000 0.2500000000000000 0.2500000000000000
CONTCAR內記載了POSCAR定義的系統經過結構最適化操作後的最終結果,而其格式與POSCAR完全相同。根據我們的計算,使用這些計算條件後所預估的晶格常數為2.734?? × 2 = 5.468A,與實驗值接近(約5.431 A)。
OSZICAR
此檔案記錄計算過程中的能量變化,而格式如下圖所示:
| |
N
|
E
|
dE
|
d eps
|
ncg
|
rms
|
rms(c)
|
| DAV: |
1 |
-0.108927449532E+02 |
-0.86129E-01 |
-0.11775E+00 |
10440 |
0.249E+00 |
0.691E-01 |
| DAV: |
2 |
-0.108726082000E+02 |
0.20137E-01 |
-0.14828E-02 |
10400 |
0.351E-01 |
0.394E-01 |
| DAV: |
3 |
-0.108624927589E+02 |
0.10115E-01 |
-0.22498E-02 |
11620 |
0.316E-01 |
0.522E-02 |
| DAV: |
4 |
-0.108622973873E+02 |
0.19537E-03 |
-0.80674E-04 |
8800 |
0.169E-01 |
0.149E-02 |
| DAV: |
5 |
-0.108624779098E+02 |
-0.18052E-03 |
-0.70490E-04 |
8800 |
0.175E-01 |
0.565E-03 |
| DAV: |
6 |
-0.108625497167E+02 |
-0.71807E-04 |
-0.87128E-05 |
8880 |
0.627E-02 |
|
3 F= -.10862550E+02 E0= -.10862671E+02 d E =-.106254E+00 mag= 0.0000
上方的DAV:1至DAV:6代表電子進行自洽計算時其能量的演變,而當自洽計算達到收斂條件時即會在下方「3 F=」之行中顯示這次計算後所求得之系統總能(不含電子entropy的F及含電子entropy approximation的E)、能量的收斂狀況(dE)、以及自旋總量(mag)。
OUTCAR
此檔案中記載了所有計算相關的參數與計算過程中的系統演變,同時包含INCAR、CONTCAR和OSZICAR的內容。然而,由於這檔案內容相當龐大且繁雜,通常我們並不會直接逐行閱讀其內容,而是會使用「grep」等指令擷取需要讀取的部分。
在結構最適化中,此檔案最重要的資訊為系統承受應力(external stress)的變化。此值可藉由「grep external OUTCAR」的指令中得到。
二-3 二氧化矽(SiO2, α-quartz)的結構最適化
在此計算中,我們使用與矽相同的INCAR與KPOINT檔,而其POSCAR如下:
SiO2_quartz
1.0
4.9800
-2.49 4.31 0
0 0 5.45
O Si
6 3
Direct
0.413599998 0.267600000 0.214100003
0.732400000 0.145999998 0.547433317
0.853999972 0.586400032 0.880766690
0.267600000 0.413599998 0.785899997
0.145999998 0.732400000 0.452566653
0.586400032 0.853999972 0.119233333
0.470099986 0.000000000 0.333333343
0.000000000 0.470099986 0.666666687
0.529900014 0.529900014 0.000000000
由於α-quartz為三角晶(trigonal),其a軸與b軸的夾角為120度,而在此例子中我們對a軸與b軸的初猜長度為a = b = 4.98A、c = 5.45A。
如果我們要處理含有多種元素的系統的時候,POTCAR檔必須要包含所有元素的潛勢與位置關係。建立POTCAR的步驟如下:
Step 1:在資料夾中找出矽(Si)與氧(O)的POTCAR,並分別將其命名為POTCAR_Si與POTCAR_O
Step 2:使用cat之指令合併二檔案。需要注意的是,合併檔案的順序必須要照POSCAR內的原子順序。因為我們的POSCAR中是O在前而Si在後,故必須要照O → Si的順序建立POTCAR檔。此部分的指令如下:
cat POTCAR_O POTCAR_Si> POTCAR
如此一來,POTCAR中就含有O原子與Si原子分別相對的資料,而VASP即可據此得知兩種原子分別對應到的潛勢,並對系統進行計算。 使用此POTCAR進行結構最適化後可得到以下CONTCAR:
SiO2_quartz
1.00000000000000
4.9794621096038494 0.0000000000000000 0.0000000000000000
-2.4897310548019247 4.3102691738325047 -0.0000566532155796
0.0000000000000000 -0.0000716653765339 5.4553395792207091
O Si
6 3
Direct
0.4136055536871822 0.2671819740281773 0.2142845709292752
0.7327729428620803 0.1463788988295011 0.5475916772939201
0.8535651879010500 0.5863853761640793 0.8809450688270126
0.2671798717369757 0.4136146538359232 0.7857216181729902
0.1464235796590049 0.7328180259718228 0.4523820850707216
0.5863940740325816 0.8536210711704963 0.1190749727060762
0.4697747594032572 0.0000000000000000 0.3333333429999996
0.0000243763304126 0.4697647219429570 0.6667106277466053
0.5302
根據以上CONTCAR,藉由我們的計算我們預測α-quartz的lattice constant分別為a = b = 4.98A、c = 5.46 A。此值比實驗值(a = b = 4.92A、c = 5.41 A) 略大,但仍在合理的預測範圍內。
二-4 計算注意事項:POTCAR的選用
在任何原子模擬計算中,估計原子間的作用力永遠是最重要的過程,而在VASP的計算中POTCAR內的資訊直接影響到作用力的計算結果。因此,針對不同計算選用合適的POTCAR相當重要,而如果選擇了不適合的POTCAR即有可能得到錯誤的結果。其中,鐵的結構預測即為相當有名的例證。
根據鐵的相圖,常溫下的鐵最穩定結構為帶有磁性的BCC(體心立方)結構。然而,如果我們在計算時使用PAW-LDA的POTCAR進行結構預測,我們將未發現在常溫下的不具磁性的FCC(面心立方)結構相較帶有磁性的BCC結構較為穩定(如下圖1),而此趨勢明顯是錯誤的。反之,若我們使用GGA即可預測正確趨勢(如下圖2)。
Lattice constant vs. internal energy for BCC and FCC Fe
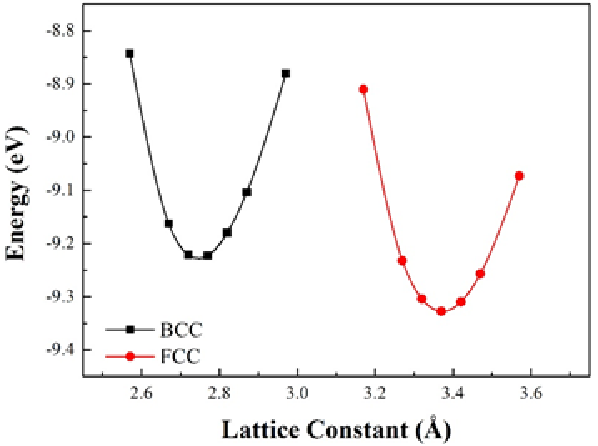
圖1:使用LDA作為exchange-correlation energy之預測
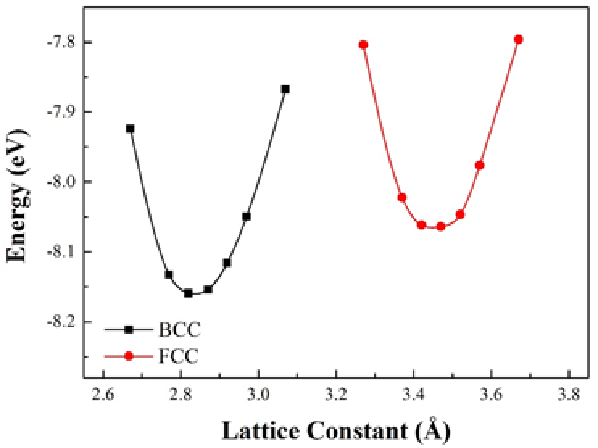
圖2:使用GGA作為exchange-correlation energy之預測
三、結語與展望
在本次的電子報中,我們示範在h71上進行VASP計算的基本方法,並實際演示VASP如何將材料的結構進行最適化,同時也強調適當選用POTCAR的重要性。這些計算是VASP最根本的操作,也是在材料計算學上不可或缺的步驟。
下次的電子報中,我們將會以此為基礎,示範如何運用VASP進行材料的性質計算,包含能帶、能量態密度等電子性質,以及材料的機械性質與振動頻譜之預測。